¿Qué es la acromegalia?
La hormona de crecimiento (GH) es una sustancia proteica liberada por la glándula pituitaria. La misma es responsable del crecimiento y desarrollo de los órganos y tejidos. La alteración en la concentración de la GH puede manifestarse en forma de acromegalia o gigantismo. Conoce más sobre esta patología a continuación.
La acromegalia es una enfermedad endocrina metabólica caracterizada por un exceso en la liberación de hormona de crecimiento. Los niveles elevados de GH en la infancia promueven el desarrollo de gigantismo. Estudios afirman que la acromegalia presenta una prevalencia mundial de 60 casos por cada millón de habitantes.
Por lo general, esta afección no varía en cuanto al sexo, la raza o la etnia. La sintomatología suele iniciar a partir de los 30 años, no obstante, la edad promedio de diagnóstico ronda los 40 años, debido a la baja sospecha de esta enfermedad. El tratamiento oportuno reduce el riesgo de complicaciones y mejora la calidad de vida.
Síntomas de la acromegalia
La acromegalia es una patología crónica y progresiva que inicia luego del cierre de las placas de crecimiento en los huesos largos. En este sentido, el exceso de GH promueve el desarrollo anormal de los tejidos blandos y la alteración de los rasgos faciales. Algunos de los signos que se evidencian en la acromegalia son los siguientes:
- Ensanchamiento de la nariz.
- Protrusión de la mandíbula.
- Abultamiento de la frente.
- Pómulos prominentes.
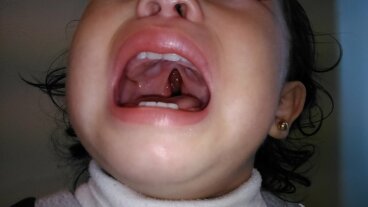
- Separación de los dientes.
- Aumento en el tamaño de manos y pies.
- Aumento en el volumen de la lengua o macroglosia.
De igual forma, es común evidenciar una piel áspera, oleosa y gruesa, así como una sudoración excesiva en las personas afectadas. Otras de las manifestaciones generales de esta patología son las siguientes:
- Voz de tono áspero y grave.
- Fatiga y debilidad muscular.
- Ronquidos.
- Dolor de cabeza persistente.
- Problemas visuales.
- Movimiento articular limitado.
Causas

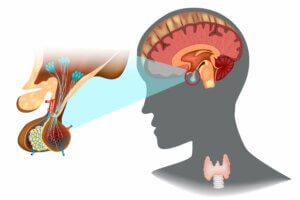
La hipófisis o glándula pituitaria es un pequeño órgano neuroendocrino ubicado en la base del cráneo. Este se encarga de secretar diversas hormonas y regular el funcionamiento de diversos procesos como la ovulación. La GH se sintetiza y libera desde las células somatótropas de la pituitaria.
La acromegalia es resultado de la producción y liberación excesiva de GH en los adultos de forma crónica. Esta hormona es capaz de aumentar la síntesis de proteínas y el crecimiento de los tejidos. Además, promueve la secreción del factor de crecimiento similar a la insulina tipo 1 (IGF-1), responsable del desarrollo del tejido blando y los huesos.
Investigaciones afirman que más del 98 % de los casos de acromegalia son resultado de un adenoma hipofisario. Este es un tipo de tumor benigno capaz de comprimir a las estructuras hipofisarias, aumentando la producción de GH y otras hormonas. De igual forma, la compresión tumoral también origina dolor de cabeza y alteraciones visuales.
Por otro lado, la acromegalia también es el resultado de tumores secretores de GH ubicados en el pulmón o el páncreas. Además, este tipo de tumores tienen la capacidad de sintetizar la hormona liberadora de hormona de crecimiento (GHRH). Esta molécula activa la liberación GH directamente en la hipófisis.
Complicaciones de la acromegalia
Los niveles elevados de GH en el organismo pueden traer graves consecuencias a largo plazo. Las complicaciones cardiovasculares afectan a la mitad de las personas con acromegalia. La hipertensión arterial, la cardiopatía hipertrófica, las arritmias y la enfermedad coronaria son algunas de las complicaciones más comunes.
Por otro lado, las personas afectadas pueden desarrollar con el tiempo diversas afecciones pulmonares, articulares, neurológicas y endocrino metabólicas. Dentro de las principales complicaciones de la acromegalia se encuentran las siguientes:
- Apnea obstructiva del sueño e insuficiencia respiratoria.
- Dolores articulares y fracturas vertebrales.
- Síndrome de túnel carpiano.
- Agrandamiento de la tiroides.
- Pólipos colónicos y cáncer colorrectal.
- Diabetes tipo 2.
- Aumento de los triglicéridos.
De igual forma, algunos pacientes pueden manifestar alteraciones de la esfera psicológica. Las mismas incluyen disminución de la libido, ansiedad, depresión y demencia. La falta de tratamiento oportuno pueden promover un deterioro en la calidad de vida y la muerte prematura.
Diagnóstico
El diagnóstico de la acromegalia se basa en la detección de las manifestaciones clásicas de la enfermedad y su confirmación por estudios complementarios. En este sentido, el médico debe considerar los antecedentes personales y familiares de la persona, así como los hallazgos a la exploración física.
De igual forma, la medición en sangre de los niveles de GH e IGF-1 constituyen la base para el diagnóstico de esta afección. La concentración de GH en adultos normales es casi indetectable. Por su parte, un valor de IGF-1 entre 122 y 400 nanogramos por mililitro excluye a la acromegalia.
Algunos estudios sugieren que la prueba de supresión de GH con carga oral de glucosa es el estándar de oro para la identificación de esta entidad. En la misma se miden los niveles de GH antes y después de la administración de 75 gramos de glucosa. Resulta normal cuando se detecta una supresión de la GH por debajo de 1 nanogramo por mililitro.
En ocasiones es necesario medir la proteína transportadora 3 del IGF-1 y los niveles de hormona liberadora de hormona de crecimiento. Además, es útil la realización de otros análisis complementarios como los siguientes:
- Niveles de azúcar en sangre.
- Prueba de prolactina.
- Tomografía axial computarizada (TAC).
- Resonancia magnética nuclear (RMN).
- Ecocardiografía y colonoscopia.
Tratamiento
El objetivo principal del tratamiento de la acromegalia es lograr la remisión de los síntomas y controlar la lesión tumoral sin alterar el funcionamiento de la hipófisis. De igual forma, se busca reducir los niveles de GH por debajo de 1 nanogramo por mililitro en la prueba de carga oral de glucosa.
El plan terapéutico varía según la gravedad del cuadro clínico, la edad y el estado general del paciente, así como la localización y el tamaño de tumor. Dentro de las opciones de tratamiento se incluyen la cirugía y la radioterapia para reducir la masa tumoral, así como los medicamentos para disminuir los niveles de GH.
Cirugía

Constituye la primera línea terapéutica en la mayoría de las personas con acromegalia. La extirpación tumoral permite que los niveles de GH vuelvan a la normalidad y reduce la sintomatología asociada a la compresión de los tejidos adyacentes.
La endoscopia transesfenoidal es el método más utilizado para la extracción de las lesiones hipofisarias. La congestión nasal, la sinusitis y el sangrado nasal son algunas de las complicaciones asociadas a este procedimiento. No obstante, la misma tiene un alto porcentaje de éxito y la tasa de mortalidad es muy reducida.
Medicamentos
El uso de fármacos está indicado en aquellos pacientes que no son candidatos a cirugía o en las personas que persista la enfermedad luego del acto quirúrgico. Los medicamentos más empleados en la acromegalia son los siguientes:
- Agonistas de la somatostatina: actúan reduciendo la síntesis y liberación de GH en la glándula pituitaria. El octreótido o Sandostatin ®, y el lanreótido o Somatuline Depot ® son algunas de las formulaciones usadas. Por lo general, se administran por vía intramuscular o subcutánea.
- Antagonistas de la hormona de crecimiento: son análogos sintéticos de la GH que bloquean los receptores específicos de esta sustancia y evitan su activación. El pegvisomant o Somavert es el principal representante de este grupo. El mismo se administra por vía subcutánea y reduce los niveles de IGF-1.
- Agonistas de la dopamina: son fármacos capaces de reducir las concentraciones de GH e IGF-1. La cabergolina y la bromocriptina son drogas de administración oral a dosis altas capaces de reducir el tamaño de las lesiones tumorales. Suelen utilizarse solas o en combinación con cualquiera de los medicamentos anteriores.
Radioterapia
La radioterapia es empleada como terapia adyuvante en las personas en las que persiste la enfermedad luego de la cirugía. Además, es útil cuando los medicamentos no ofrecen buenos resultados o están contraindicados. La misma actúa destruyendo las células tumorales y por consiguiente disminuyendo los niveles de GH.
Los métodos de radioterapia incluyen la forma convencional y estereotáctica. Esta última es la más recomendada, ya que se aplica una única dosis y tiene menos efectos adversos.
Una afección que debe ser detectada a tiempo
La acromegalia es una enfermedad poco frecuente asociada a niveles elevados de GH en los adultos. Los síntomas característicos incluyen las alteraciones en los rasgos faciales y el crecimiento anormal de manos y pies. La falta de tratamiento puede promover varias complicaciones a largo plazo que ponen en riesgo la vida de las personas.
Ante la evidencia de los síntomas clásicos de esta afección es vital buscar atención médica. Los profesionales de salud son los únicos capacitados para valorar esta enfermedad y ofrecer la mejor guía en el alivio de los síntomas.
La hormona de crecimiento (GH) es una sustancia proteica liberada por la glándula pituitaria. La misma es responsable del crecimiento y desarrollo de los órganos y tejidos. La alteración en la concentración de la GH puede manifestarse en forma de acromegalia o gigantismo. Conoce más sobre esta patología a continuación.
La acromegalia es una enfermedad endocrina metabólica caracterizada por un exceso en la liberación de hormona de crecimiento. Los niveles elevados de GH en la infancia promueven el desarrollo de gigantismo. Estudios afirman que la acromegalia presenta una prevalencia mundial de 60 casos por cada millón de habitantes.
Por lo general, esta afección no varía en cuanto al sexo, la raza o la etnia. La sintomatología suele iniciar a partir de los 30 años, no obstante, la edad promedio de diagnóstico ronda los 40 años, debido a la baja sospecha de esta enfermedad. El tratamiento oportuno reduce el riesgo de complicaciones y mejora la calidad de vida.
Síntomas de la acromegalia
La acromegalia es una patología crónica y progresiva que inicia luego del cierre de las placas de crecimiento en los huesos largos. En este sentido, el exceso de GH promueve el desarrollo anormal de los tejidos blandos y la alteración de los rasgos faciales. Algunos de los signos que se evidencian en la acromegalia son los siguientes:
- Ensanchamiento de la nariz.
- Protrusión de la mandíbula.
- Abultamiento de la frente.
- Pómulos prominentes.
- Separación de los dientes.
- Aumento en el tamaño de manos y pies.
- Aumento en el volumen de la lengua o macroglosia.
De igual forma, es común evidenciar una piel áspera, oleosa y gruesa, así como una sudoración excesiva en las personas afectadas. Otras de las manifestaciones generales de esta patología son las siguientes:
- Voz de tono áspero y grave.
- Fatiga y debilidad muscular.
- Ronquidos.
- Dolor de cabeza persistente.
- Problemas visuales.
- Movimiento articular limitado.
Causas
La hipófisis o glándula pituitaria es un pequeño órgano neuroendocrino ubicado en la base del cráneo. Este se encarga de secretar diversas hormonas y regular el funcionamiento de diversos procesos como la ovulación. La GH se sintetiza y libera desde las células somatótropas de la pituitaria.
La acromegalia es resultado de la producción y liberación excesiva de GH en los adultos de forma crónica. Esta hormona es capaz de aumentar la síntesis de proteínas y el crecimiento de los tejidos. Además, promueve la secreción del factor de crecimiento similar a la insulina tipo 1 (IGF-1), responsable del desarrollo del tejido blando y los huesos.
Investigaciones afirman que más del 98 % de los casos de acromegalia son resultado de un adenoma hipofisario. Este es un tipo de tumor benigno capaz de comprimir a las estructuras hipofisarias, aumentando la producción de GH y otras hormonas. De igual forma, la compresión tumoral también origina dolor de cabeza y alteraciones visuales.
Por otro lado, la acromegalia también es el resultado de tumores secretores de GH ubicados en el pulmón o el páncreas. Además, este tipo de tumores tienen la capacidad de sintetizar la hormona liberadora de hormona de crecimiento (GHRH). Esta molécula activa la liberación GH directamente en la hipófisis.
Complicaciones de la acromegalia
Los niveles elevados de GH en el organismo pueden traer graves consecuencias a largo plazo. Las complicaciones cardiovasculares afectan a la mitad de las personas con acromegalia. La hipertensión arterial, la cardiopatía hipertrófica, las arritmias y la enfermedad coronaria son algunas de las complicaciones más comunes.
Por otro lado, las personas afectadas pueden desarrollar con el tiempo diversas afecciones pulmonares, articulares, neurológicas y endocrino metabólicas. Dentro de las principales complicaciones de la acromegalia se encuentran las siguientes:
- Apnea obstructiva del sueño e insuficiencia respiratoria.
- Dolores articulares y fracturas vertebrales.
- Síndrome de túnel carpiano.
- Agrandamiento de la tiroides.
- Pólipos colónicos y cáncer colorrectal.
- Diabetes tipo 2.
- Aumento de los triglicéridos.
De igual forma, algunos pacientes pueden manifestar alteraciones de la esfera psicológica. Las mismas incluyen disminución de la libido, ansiedad, depresión y demencia. La falta de tratamiento oportuno pueden promover un deterioro en la calidad de vida y la muerte prematura.
Diagnóstico
El diagnóstico de la acromegalia se basa en la detección de las manifestaciones clásicas de la enfermedad y su confirmación por estudios complementarios. En este sentido, el médico debe considerar los antecedentes personales y familiares de la persona, así como los hallazgos a la exploración física.
De igual forma, la medición en sangre de los niveles de GH e IGF-1 constituyen la base para el diagnóstico de esta afección. La concentración de GH en adultos normales es casi indetectable. Por su parte, un valor de IGF-1 entre 122 y 400 nanogramos por mililitro excluye a la acromegalia.
Algunos estudios sugieren que la prueba de supresión de GH con carga oral de glucosa es el estándar de oro para la identificación de esta entidad. En la misma se miden los niveles de GH antes y después de la administración de 75 gramos de glucosa. Resulta normal cuando se detecta una supresión de la GH por debajo de 1 nanogramo por mililitro.
En ocasiones es necesario medir la proteína transportadora 3 del IGF-1 y los niveles de hormona liberadora de hormona de crecimiento. Además, es útil la realización de otros análisis complementarios como los siguientes:
- Niveles de azúcar en sangre.
- Prueba de prolactina.
- Tomografía axial computarizada (TAC).
- Resonancia magnética nuclear (RMN).
- Ecocardiografía y colonoscopia.
Tratamiento
El objetivo principal del tratamiento de la acromegalia es lograr la remisión de los síntomas y controlar la lesión tumoral sin alterar el funcionamiento de la hipófisis. De igual forma, se busca reducir los niveles de GH por debajo de 1 nanogramo por mililitro en la prueba de carga oral de glucosa.
El plan terapéutico varía según la gravedad del cuadro clínico, la edad y el estado general del paciente, así como la localización y el tamaño de tumor. Dentro de las opciones de tratamiento se incluyen la cirugía y la radioterapia para reducir la masa tumoral, así como los medicamentos para disminuir los niveles de GH.
Cirugía
Constituye la primera línea terapéutica en la mayoría de las personas con acromegalia. La extirpación tumoral permite que los niveles de GH vuelvan a la normalidad y reduce la sintomatología asociada a la compresión de los tejidos adyacentes.
La endoscopia transesfenoidal es el método más utilizado para la extracción de las lesiones hipofisarias. La congestión nasal, la sinusitis y el sangrado nasal son algunas de las complicaciones asociadas a este procedimiento. No obstante, la misma tiene un alto porcentaje de éxito y la tasa de mortalidad es muy reducida.
Medicamentos
El uso de fármacos está indicado en aquellos pacientes que no son candidatos a cirugía o en las personas que persista la enfermedad luego del acto quirúrgico. Los medicamentos más empleados en la acromegalia son los siguientes:
- Agonistas de la somatostatina: actúan reduciendo la síntesis y liberación de GH en la glándula pituitaria. El octreótido o Sandostatin ®, y el lanreótido o Somatuline Depot ® son algunas de las formulaciones usadas. Por lo general, se administran por vía intramuscular o subcutánea.
- Antagonistas de la hormona de crecimiento: son análogos sintéticos de la GH que bloquean los receptores específicos de esta sustancia y evitan su activación. El pegvisomant o Somavert es el principal representante de este grupo. El mismo se administra por vía subcutánea y reduce los niveles de IGF-1.
- Agonistas de la dopamina: son fármacos capaces de reducir las concentraciones de GH e IGF-1. La cabergolina y la bromocriptina son drogas de administración oral a dosis altas capaces de reducir el tamaño de las lesiones tumorales. Suelen utilizarse solas o en combinación con cualquiera de los medicamentos anteriores.
Radioterapia
La radioterapia es empleada como terapia adyuvante en las personas en las que persiste la enfermedad luego de la cirugía. Además, es útil cuando los medicamentos no ofrecen buenos resultados o están contraindicados. La misma actúa destruyendo las células tumorales y por consiguiente disminuyendo los niveles de GH.
Los métodos de radioterapia incluyen la forma convencional y estereotáctica. Esta última es la más recomendada, ya que se aplica una única dosis y tiene menos efectos adversos.
Una afección que debe ser detectada a tiempo
La acromegalia es una enfermedad poco frecuente asociada a niveles elevados de GH en los adultos. Los síntomas característicos incluyen las alteraciones en los rasgos faciales y el crecimiento anormal de manos y pies. La falta de tratamiento puede promover varias complicaciones a largo plazo que ponen en riesgo la vida de las personas.
Ante la evidencia de los síntomas clásicos de esta afección es vital buscar atención médica. Los profesionales de salud son los únicos capacitados para valorar esta enfermedad y ofrecer la mejor guía en el alivio de los síntomas.
- Rojas-Zalazar D, Mura C J, Cataldo G C, Wohllk G N. Manejo multidisciplinario de la acromegalia. Rev. chil. neuro-psiquiatr. 2011 Mar ; 49( 1 ): 37-46.
- Mercano M. Complicaciones sistémicas de la acromegalia. Rev. Venez. Endocrinol. Metab. 2006 Oct; 4( 3 ): 006-006.
- González-Houdelath K. Acromegalia. Revista Medica Sinergia. 2020;5(7):e540.
- Chanson P, Salenave S. Acromegaly. Orphanet J Rare Dis. 2008 Jun 25;3:17.
- Melmed S. Acromegaly pathogenesis and treatment. J Clin Invest. 2009 Nov;119(11):3189-202.
- Lavrentaki A, Paluzzi A, Wass JA, Karavitaki N. Epidemiology of acromegaly: review of population studies. Pituitary. 2017 Feb;20(1):4-9.
Este texto se ofrece únicamente con propósitos informativos y no reemplaza la consulta con un profesional. Ante dudas, consulta a tu especialista.